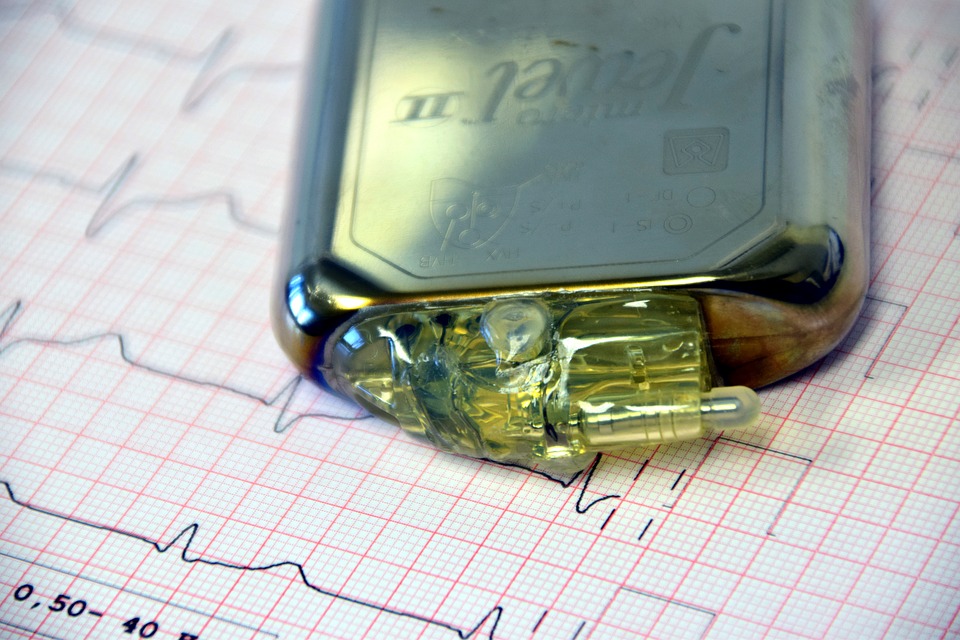
According to an investigation conducted by the Kaiser Health News, the Food and Drug Administration (FDA) allowed Medtronic, a company who manufactured an implantable heart device called Sprint Fidelis, to secretly log 50,000 malfunction incidents.
The device in question is made out of a few wires and a defibrillator and its main purpose is to send the heart back into its regular rhythm through a series of jolts, if it senses something is wrong.
However, according to some doctors, the Sprint Fidelis was giving patients random and harmful jolts without reason while other times, during actual cardiac emergencies, it failed to function altogether.
The device was implanted in 268,000 patients before Medtronic took the decision to recall the device in 2007. Of course, following the recall, there was only so much the patients could do: some of them had to find ways to live with the device while others decided to undergo risky surgeries to remove it.
Dr John Mandrola, cardiologist at the Baptist Medical Associates in Louisville, Ky. has stated in the investigative report that the Sprint Fidelis has traumatized some of his patients thanks to the unexpected shocks and that others had ended up suffering repercussions from the faulty device for years.
He went on to say that, the fact that the FDA did not release the reports to the public about a device that continued to harm its users seemed “problematic” to him. “What is the benefit to the public of an exemption?” he went on to ask.
Dr. Frederic Resnic, cardiovascular medicine division chairman at the Lahey Hospital & Medical Center in Burlington, Mass. also added in a written email to KHN that the “lack of communication and transparency challenged the FDA’s unique role as the primary, trusted, information source regarding medical service safety.”
Reports of malfunctioning devices such as the Spring Fidelis are recorded in an FDA database called Manufacturer and User Facility Device Experience (MAUDE) but, according to the investigation, the FDA and Medtronic “made a deal to keep reports about the widely used device’s malfunction incidents“, which totaled around 50,000 reports that were kept away from the public view.
Apparently, the agency created an “alternative summary reporting” repository in order to cut down on paperwork for redundant incident reports but most doctors, consumer advocates and even the higher-ranking FDA employees were completely in the dark about its existence.
Following the investigative report from KHN, the FDA announced that it’s shutting down the “alternative summary reporting” repository in a move that former FDA official Lori S. Brown describes as a “victory for patients and consumers”.
If you or someone you know have concerns about the safety of your or their medical devices, you can contact the government agencies to self-report any issue, over at this link.
Follow TechTheLead on Google News to get the news first.